Aligning to a reference sequence when you have treated your sample with Sodium bisulfite is no easy task. Global or "end-to-end" read aligners are generally very susceptible to sequence variants. During bisulfite conversion, all cytosines (C) will be converted to a Uracil (U), while 5' methyl cytosine's will be left unconverted as a cytosine (C). During amplification the converted Uracil's (U) are read as Thymine's (T).
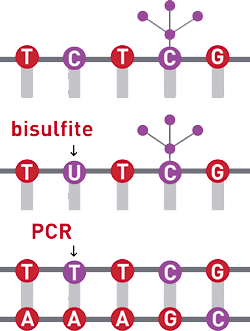
The consequence is that the conversion introduces a significant amount of sequence diversity which impacts genome alignment.
However, there are a number of programs that use existing global read aligners, such as bwa and bowtie(1/2), by aligning reads to converted and non-converted reference genomes.
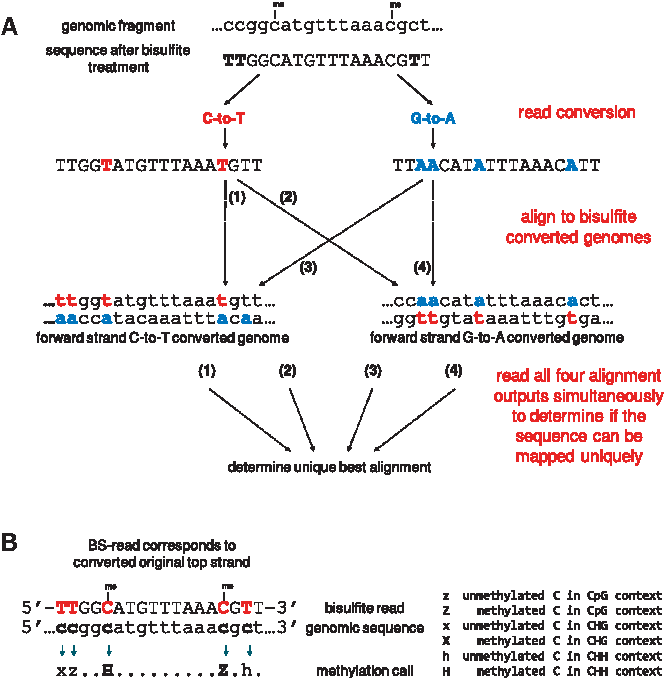
The mapping procedure and program that we will be using today is the bismark WGBS program, which uses the bowtie2 alignment tool to align reads.
As shown above, reads are aligned to a reference genome that is converted into two different bowtie2 indexes:
- a reference sequence that converts all the C's to T's
- a reference sequence that converts all the G's to A's (i.e. C->T on the opposite strand)
With our reduced reference genome of Arabidopsis_thaliana, we now need to prepare our genome for alignment.
Move the reference genome sequence (thats in a compressed format "fa.gz") into a new directory and use the bismark_genome_preparation command to run all the bowtie2 indexing.
# Create directory for bismark WGBS genome
$ mkdir Athal
# Move data file in there
$ mv TAIR10_chr1_cp.fa.gz Athal/
# Run bismark to format our Athaliana genome (TAIR10)
$ bismark_genome_preparation --bowtie2 Athal
Now we have all of the index sequences we need to map our WGBS reads to Arabidopsis_thaliana.
To demonstrate all the files bismark_genome_preparation has made, lets list all of the files found within the new directory.
$ ls -R Athal
Questions
- How many
bowtie2index files are created for each converted sequence?
Now that we have a bismark formatted reference sequence and our trimmed FASTQ files, we can now go straight into aligning the data.
We have single end sequencing data, meaning that all we need to specify is our sequence files, the bismark reference directory (Athal), the aligner that we want to use or that our reference sequence is formatted for (--bowtie2), the ouput type we want to produce (--bam) and the number of threads we'd like to use (-p 2).
# Map SRR534177
$ bismark -p 2 --bam --bowtie2 Athal SRR534177_colWT_trimmed.fq.gz
# Map SRR534239
$ bismark -p 2 --bam --bowtie2 Athal SRR534239_met1_trimmed.fq.gz
These should take ~5-20mins to complete per sample so dont worry if they are taking their time. At the end of the process you should have the following files:
$ ls -l
To look at the
$ less SRR534177_colWT_trimmed_bismark_bt2_SE_report.txt
The alignment file that we produced is called a Binary Alignment Map or BAM file. This is actually just the binary compressed version of a SAM file (Sequence Alignment Map), so SAM and BAM files are often used interchangably for a lot of genomics applications. SAM files are text based files, whereas BAMs are just the binary version of that text. BAM is the main alignment file format of choice for most applications because of the storage saving you get from having a compressed file. Depending on the compression rate used, a BAM file is often up to 10x smaller that the SAM equivalent, which is a major saving on space when dealing with 30x coverage whole genomes!! To make things even more confusing, in the last 10 years a new alignment format was created called CRAM. CRAM was designed to be an efficient reference-based alternative to the Sequence Alignment Map (SAM) and Binary Alignment Map (BAM) file formats.
Each of these alignment formats present alignment information that is column-based (much like a basic spreadsheet) with rows containing each read from the input FASTQ file. The columns for this file are as follows:
| Col | Field | Type | Brief description |
|---|---|---|---|
| 1 | QNAME | String | Query template NAME |
| 2 | FLAG | Int | bitwise FLAG |
| 3 | RNAME | String | References sequence NAME |
| 4 | POS | Int | 1- based leftmost mapping POSition |
| 5 | MAPQ | Int | MAPping Quality |
| 6 | CIGAR | String | CIGAR string |
| 7 | RNEXT | String | Ref. name of the mate/next read |
| 8 | PNEXT | Int | Position of the mate/next read |
| 9 | TLEN | Int | observed Template LENgth |
| 10 | SEQ | String | segment SEQuence |
| 11 | QUAL | String | ASCII of Phred-scaled base QUALity+33 |
For more information on BAM format, make sure you check out the University of Adelaide Bioinformatics Hub's "Intro to NGS" tutorial.
Lets have a look at our brand new alignment file. Firstly, lets look at the header:
$ samtools view -H SRR534177_colWT_trimmed_bismark_bt2.bam
@HD VN:1.0 SO:unsorted
@SQ SN:1 LN:30427671
@SQ SN:chloroplast LN:154478
@PG ID:Bismark VN:v0.22.1 CL:"bismark -p 8 --bam --bowtie2 Athalchr1cp SRR534177_colWT_trimmed.fq.gz"
Now lets have a look at the rest of the BAM file. Because there is a lot of lines in this file, lets just look at the top portion:
$ samtools view SRR534177_colWT_trimmed_bismark_bt2.bam | head -n 4
SRR534177.13_SN603_WA034:3:1101:45.50:110.90/1 16 1 10011165 42 40M * 0 0 AATCGCATATTCACATACATATAATAAAATTTTATAAAAT GHE?4>HBCHBHBBHFFEEBHAIIIGIIIIHBFBFFHDDD NM:i:7 MD:Z:0G3A11G6G4G4G1G4 XM:Z:h...............h......h....h....h.h.... XR:Z:CT XG:Z:GA
SRR534177.18_SN603_WA034:3:1101:42.80:117.20/1 16 1 17342024 42 40M * 0 0 TACTCTAAAAATTATACCATTATTTTGTTAAATCCGTACC FBIIIIJJJJIIGF?9GGEGGFIGGHBGGHGGFHHFD?FD NM:i:8 MD:Z:1G4G1G6G5G7G0G6G2 XM:Z:.x....x.h......h.....h....H..hh....Z.h.. XR:Z:CT XG:Z:GA
SRR534177.34_SN603_WA034:3:1101:51.50:109.90/1 16 1 19411065 42 39M * 0 0 CAAATATAAATTCCGCACAAATTCCTACAACATTTTCAT JJJJJJIJJJGGJIIIHJJJJIGIHGDJIIJHHHHHFFF NM:i:4 MD:Z:9G9G0G8G9 XM:Z:.........h....Z....xh........x......... XR:Z:CT XG:Z:GA
SRR534177.36_SN603_WA034:3:1101:72.10:110.50/1 0 chloroplast 47672 42 37M * 0 0 ATAGATTTAAGTTATATATTAAAATGATATTGATATT FFFHHHHHJJJIJJJJJJJJJJJJJJIJJJJJJJIJJ NM:i:5 MD:Z:5C0C5C16C5C1 XM:Z:.....hh.....h................x.....h. XR:Z:CT XG:Z:CT
As you can see, there is a lot of information contained in these files.
The first 11 fields are standard across SAM/BAM files, but the additional fields contain extra metadata information that is introduced by the aligner.
These bismark specific tags contain important information that can be read by methylation callers.

XR tag
- Denotes the read conversion state for the alignment at a genomic location.
- This tag with CT denotes that the read is aligned in its original form (as it is) as sequenced.
- This tag with GA denotes that the reverse compliment of the read is aligned.
XG tag
- Denotes the Genome conversion state for the alignment
- This tag with CT denotes that the read is aligned to the OT of reference.
- This tag with GA denotes that the read is aligned from the OB of reference.
XM tags
- Aligned Read list used for computation of methylation calls
- Total number of Cs analysed = sum of z, Z, x, X, h, H counts
- Total number of methylated Cs in CpG context = Z counts
- Total number of methylated Cs in CHG context = X counts
- Total number of methylated Cs in CHH context = H counts
- Total number of C to T conversions in CpG context = z counts
- Total number of C to T conversions in CHG context = x counts
- Total C to T conversions in CHH context = h counts
These call all be used by the methylation caller (see next section) to define the methylation proportion at each site and context.
The key point to understand with this file is that the output of bismark will contain ALL reads and their alignment information, regardless of whether they aligned or not.
Information about what read aligned, in what orientation etc can all be gathered from the bitwise FLAG column in the BAM file.
These FLAGs can be used to subset the BAM file to contain only the reads that you're interested in.
A really nice explainer of SAM Flags are found at this site: https://broadinstitute.github.io/picard/explain-flags.html.
These can be used with the samtools view command:
$ samtools view
Usage: samtools view [options] <in.bam>|<in.sam>|<in.cram> [region ...]
...
...
...
-f INT only include reads with all of the FLAGs in INT present [0]
-F INT only include reads with none of the FLAGS in INT present [0]
For example, I want to get all of the reads that map to the genome.
The SAM FLAG for read unmapped is FLAG 4, meaning that I want none of those to be present in my extracted reads.
So running samtools view with -F 4, should give me all my mapped reads
$ samtools view -F 4 SRR534177_colWT_trimmed_bismark_bt2.bam | wc -l
Obviously you dont want to be running through every FLAG one by one, so a nice little summary of all FLAG information can be obtained with the samtools flagstat command
$ samtools flagstat SRR534177_colWT_trimmed_bismark_bt2.bam
Questions
- How many reads mapped to the genome?
- How many reads where mapped as the second pair?
- What possible explaination might there be for the lack of reads in most of the FLAGs available in
samtools flagstat?
NEXT: We'll now start using these BAM files to call methylated sites